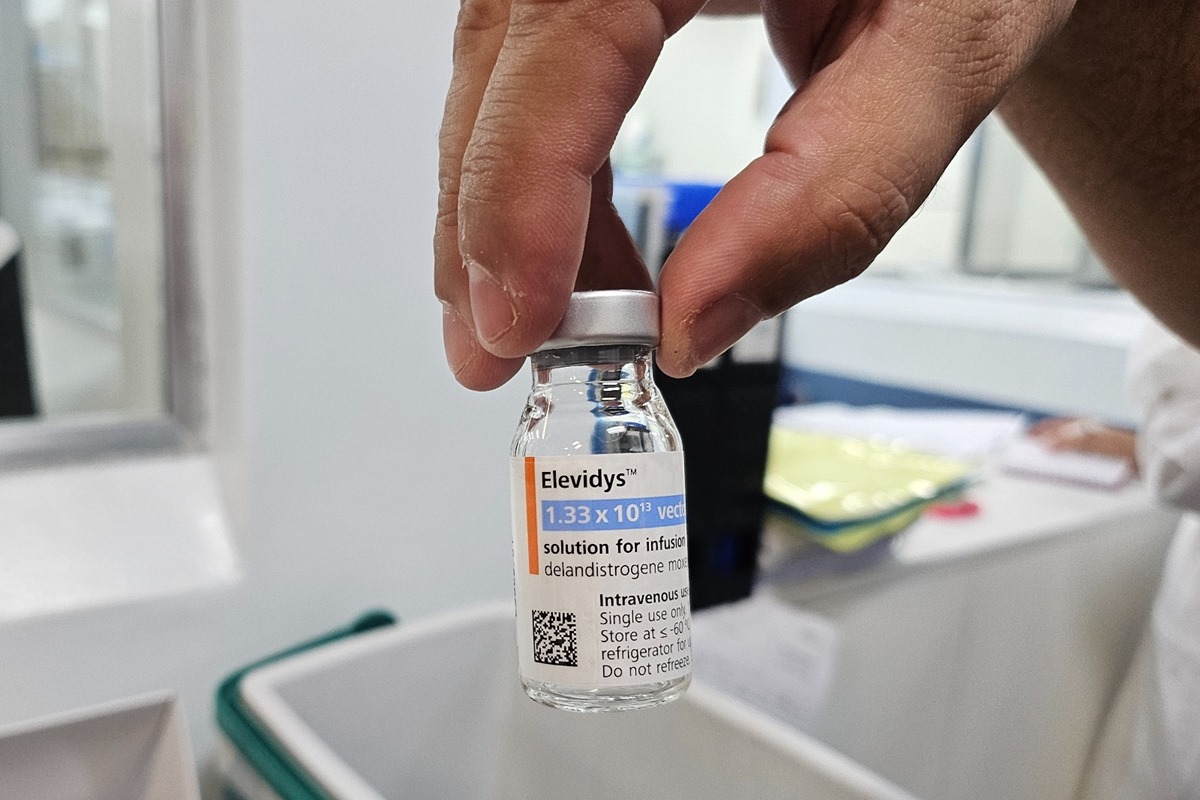
Anvisa suspende medicamento de R$ 17 milhões após mortes nos EUA
- Brasil
- 25/07/2025
- No Comment
- 12
A Agência Nacional de Vigilância Sanitária (Anvisa) suspendeu temporariamente, por medida de precaução, a comercialização, distribuição, fabricação importação, propaganda e uso do medicamento de terapia gênica Elevidys (delandistrogeno moxeparvoveque) no Brasil.
Segundo a agência, a suspensão, que foi publicada em edição extra do Diário Oficial dessa quinta-feira (24/7), vale até que sejam integralmente esclarecidas as recentes incertezas de segurança relacionadas a relatos de casos fatais de insuficiência hepática aguda, ou seja, falência do fígado, em pacientes tratados com o produto nos Estados Unidos.
O medicamento, considerado um dos mais caros do mundo, chegando ao valor de R$ 17 milhões, é o único capaz de frear o avanço da distrofia muscular de Duchenne (DMD), uma doença genética rara e degenerativa.
Remédio mais caro do Brasil
- O remédio Elevidys é considerado um dos mais caros do mundo e o único disponível no Brasil para o tratamento de crianças com distrofia muscular de Duchenne (DMD).
- O medicamento foi aplicado pela primeira vez no Sistema Único de Saúde (SUS) em fevereiro deste ano. A Câmara de Regulação do Mercado de Medicamentos (CMED) estipulou um valor máximo de R$ 11 milhões, fazendo dele o remédio mais caro disponível no país.
- A distrofia muscular de Duchenne, ou síndrome de Duchenne, é uma doença genética rara que acomete, principalmente, pessoas do sexo masculino.
- A alteração genética é caracterizada pela falta ou alteração da proteína distrofina no músculo das crianças, que ocasiona o principal sintoma da doença: fraqueza muscular. A condição pode levar à perda progressiva de habilidades motoras, como subir escadas, pular e correr.
Óbitos
Conforme a Anvisa, a decisão da suspensão foi tomada à luz de novas informações regulatórias divulgadas pela Food and Drug Administration (FDA), agência reguladora dos Estados Unidos, que relata três óbitos associados ao uso de produtos de terapia gênica, sendo dois casos em pacientes pediátricos não deambuladores com Distrofia Muscular de Duchenne (DMD) tratados com o Elevidys e um terceiro caso em paciente adulto com Distrofia Muscular de Cinturas, que utilizaram produto de terapia gênica experimental baseado no mesmo vetor viral.
O primeiro caso ocorreu em março, quando um jovem de 16 anos faleceu após usar o remédio. A segunda vítima foi um adolescente de 15 anos. Ambos receberam a medicação no momento em que não conseguiam mais andar devido ao agravamento da doença, o que caracteriza o uso off-label.
Os eventos envolveram evolução para insuficiência hepática aguda com desfecho fatal. Segundo a agência é importante destacar que, nos Estados Unidos, a empresa Sarepta Therapeutics é a responsável pelo registro do Elevydis. Já no Brasil, o registro do medicamento foi concedido à Roche Farma Brasil em dezembro de 2024.
Uso no Brasil
No Brasil, o uso do Elevidys é indicado apenas para crianças com DMD com idades entre 4 e 7 anos que ainda conseguem andar. Em comunicado, a Anvisa afirmou que a restrição vigente no país já exclui os pacientes que não conseguem caminhar, como os que morreram nos EUA.
Embora os casos de óbito tenham ocorrido em perfil de pacientes para os quais o Elevidys não está indicado no Brasil e não haja, até o momento, relatos de problemas relacionados à sua aplicação no país, a Anvisa decidiu suspender temporariamente o uso do produto.
Segundo a Anvisa, a decisão foi tomada de forma cientificamente prudente e preventiva, diante do surgimento de novos dados internacionais que apontam para hepatotoxicidade grave com desfecho fatal. Desta forma, apesar da situação restritiva e controlada do uso no Brasil e da experiência internacional com mais de 700 pacientes tratados sem óbitos na população deambuladora, a suspensão temporária do uso do produto, como medida preventiva, em alinhamento com a empresa Roche, tem o objetivo de:
- Garantir a segurança dos pacientes brasileiros, que deve sempre prevalecer sobre qualquer outra consideração;
- Permitir a avaliação aprofundada dos sinais de risco hepático emergentes nos Estados Unidos;
- Discutir, com base nos novos achados e correlações, possíveis aprimoramentos e ajustes no plano de gerenciamento de risco e nas estratégias de manejo clínico, se necessário, antes de eventual retomada da comercialização.
- Como se trata de um produto inovador, complexo e de uso restrito, qualquer dúvida razoável sobre seu perfil de segurança exige ação regulatória imediata.





 Online Agora : 0
Online Agora : 0 Seu IP : 216.73.216.13
Seu IP : 216.73.216.13